



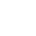




 Foto: Instituto VIDAS RARAS
Foto: Instituto VIDAS RARAS O Ministério da Saúde, por meio da Comissão Nacional de Incorporação de Tecnologias no Sistema Único de Saúde (Conitec), recomendou, em seu relatório preliminar, a não incorporação do único medicamento para a mucopolissacaridose do tipo VII (MPS VII). Porém, até o dia 11 de março, toda a sociedade poderá contribuir com o tema por meio da Consulta Pública N°3. Para participar e opinar sobre o oferecimento da medicação na rede pública de saúde, pacientes, familiares, cuidadores e cidadãos em geral devem acessar o formulário de experiência ou opinião e deixar a sua contribuição, já os médicos podem contribuir por meio do formulário técnico-científico.
Para a Conitec, a recomendação inicial pela não incorporação do alfavestronidase, única terapia de reposição enzimática disponível para esse tipo da doença no mundo para o tratamento de MPS VII em pacientes de todas as idades, se baseou no tempo de mercado e de experiência clínica com o medicamento, considerados incipientes ainda. O órgão também ressaltou o impacto orçamentário da ordem de R$ 3 milhões por paciente por ano.
Regina Próspero, presidente do Instituto Vidas Raras, afirma que justamente por ser um medicamento órfão, único para esse tipo da doença, a avaliação deveria ser distinta e considerar o pequeno número de pacientes, o tratamento atual disponível e o benefício real que essa nova terapia poderia trazer para a vida de todos que convivem com a MPS VII.
“A Conitec explica, no documento, que o tratamento atual da mucopolissacaridose tipo VII é baseado apenas em terapias de suporte e tratamentos sintomáticos, como cirurgias (reparo da hérnia, prótese de quadril e fusão cervical), antibióticos e fisioterapia. Se temos como dar acesso a um medicamento único que possa diminuir essas necessidades, de algo invasivo, por exemplo, e que realmente melhora a qualidade de vida do paciente – o que impacta também na vida de toda a família – por que negar essa possibilidade? Pacientes com outros tipos de MPS, já contam com as terapias similares no SUS. Por que essa discriminação?”, questiona.
No Brasil, no congresso da Sociedade Brasileira de Genética Médica em 2019, foi apresentado um estudo que identificou 22 casos de MPS VII no período de 15 anos. Outra pesquisa revelou que a doença atinge menos de três pessoas em cada dez milhões de nascidos vivos no país. Estima-se que sejam registrados menos de 200 casos de MPS VII em todo o mundo[2].
“São poucos os afetados diretamente por essa doença. No Brasil, atualmente estão identificados em torno de 13 pacientes. Temos que promover uma vida melhor para eles e suas famílias. Essa terapia de reposição enzimática é um dos caminhos,” explica Regina. “Precisamos contar com a participação de todos na consulta pública”.
Para o geneticista e pesquisador Roberto Giugliani, professor do Departamento de Genética da Universidade Federal do Rio Grande do Sul, Diretor do Centro Colaborador da Organização Mundial da Saúde e Membro Titular da Academia Brasileira de Ciências, os estudos clínicos comprovam que o medicamento é eficiente e seguro, apesar de bastante novo.
“Somente cerca de 5% de doenças raras ou ultrarraras, como é o caso da MPS VII, possuem alguma terapia medicamentosa apropriada. A comunidade científica já reconhece que a terapia de reposição enzimática modificou substancialmente o tratamento das mucopolissacaridoses dos tipos I, II, IV e VI na última década. No caso da MPS VII, além de termos experiência longa com outros tipos da doença, os estudos clínicos específicos mostram como o medicamento age e pode melhorar a vida desses pacientes. É um grande ganho para quem convive com a doença e não deveria ser descartado”, completa.
E o relatório preliminar da Conitec já traz os elementos que comprovam o benefício final da terapia aos pacientes, sua eficiência e segurança, segundo Edson Paixão, gerente geral da Ultragenyx, empresa responsável pelo medicamento.
“Estamos comprometidos com a comunidade de MPS do Brasil e mantemos diálogo aberto com as autoridades. Os estudos clínicos apresentam resultados significativos para os pacientes, inclusive reconhecidos pela Conitec em seu relatório inicial… entendemos que a Conitec deve prezar também pela sustentabilidade do sistema de saúde e isso se reflete no seu cuidado em relação ao impacto no orçamento global. Vamos esclarecer as dúvidas apresentadas e mostrar que, na realidade, o custo é comparável aos dos demais tratamentos de doenças raras e ultrarraras”, ressalta Paixão
Saiba mais sobre as mucopolissacaridoses – MPS é o termo utilizado para denominar um grupo de doenças raras e hereditárias que se caracterizam pela falta ou deficiência de enzimas (proteínas que auxiliam as reações químicas que acontecem no organismo), importantes na degradação de determinados tipos de açúcares (os mucopolissacarídeos). O acúmulo destes açúcares leva os pacientes a apresentarem alguns distúrbios no funcionamento de seus órgãos[3].
Na mucopolissacaridose tipo VII (MPS VII), também chamada de síndrome de Sly, falta a substância responsável pela degradação dos mucopolissacarídios (também chamados glicosaminoglicanos, GAGs), o que leva ao seu acúmulo em tecidos e órgãos e causa sintomas como atraso no desenvolvimento, deficiência intelectual, baixa estatura e dificuldade de locomoção, problemas respiratórios e cardíacos e aumento do fígado e baço.
Devido a essas complicações, a expectativa de vida é bastante reduzida: cerca de 42% dos pacientes morrem até os 35 anos, além da baixa sobrevivência dos bebês afetados com as formas mais agressivas da doença, que muitas vezes nem chegam a ser diagnosticados.
O tipo VII é transmitido dos pais para filhos, ainda que os pais não manifestem a doença. Estima-se que, no mundo, estejam atualmente registrados menos de 200 casos. Já no Brasil, um estudo apresentado no Congresso da Sociedade Brasileira de Genética Médica em 2019, identificou 22 casos de MPS VII no período de 15 anos. Outra pesquisa revelou que a doença atinge menos de 3 pessoas em cada 10 milhões de nascidos vivos no país.
O diagnóstico precoce nem sempre é possível, pois é uma doença progressiva com manifestações iniciais pouco características. A doença pode ser detectada inicialmente através da análise de glicosaminoglicanos (GAGs) na urina e confirmada através da medida da atividade da enzima beta-glicuronidase no sangue, ou por meio de sequenciamento do gene causador do problema.
Matéria: Rafael Correa













 Foto: Marcello Casal Jr/ Agência Brasil
Foto: Marcello Casal Jr/ Agência Brasil 
 Divulgação
Divulgação 
 Imagem Ilustrativa | Foto: Hélio Alves/ Tribuna do Recôncavo.
Imagem Ilustrativa | Foto: Hélio Alves/ Tribuna do Recôncavo.  Foto: PM
Foto: PM  Foto: Jackson Santos
Foto: Jackson Santos  Arte ilustrativa criada por IA
Arte ilustrativa criada por IA  Imagem de Rudy and Peter Skitterians por Pixabay
Imagem de Rudy and Peter Skitterians por Pixabay  Arte ilustrativa criada por IA
Arte ilustrativa criada por IA  Reprodução/ Vídeo
Reprodução/ Vídeo  Foto: PASCOM
Foto: PASCOM  Arquivo Pessoal
Arquivo Pessoal  Foto: Telma Galino
Foto: Telma Galino  Foto: Edílson Rodrigues/ Agência Senado
Foto: Edílson Rodrigues/ Agência Senado  Imagem ilustrativa gerada por IA
Imagem ilustrativa gerada por IA  Foto: Hélio Alves/ Tribuna do Recôncavo
Foto: Hélio Alves/ Tribuna do Recôncavo  Imagem ilustrativa gerada por IA
Imagem ilustrativa gerada por IA  Foto: Internauta do Tribuna do Recôncavo
Foto: Internauta do Tribuna do Recôncavo  Arquivo: Tribuna do Recôncavo
Arquivo: Tribuna do Recôncavo  Imagem Ilustrativa | Foto: Maria do Carmo/ Tribuna do Recôncavo
Imagem Ilustrativa | Foto: Maria do Carmo/ Tribuna do Recôncavo  Arquivo | Foto: Ney Santos
Arquivo | Foto: Ney Santos  Image by Gerd Altmann from Pixabay
Image by Gerd Altmann from Pixabay  Foto: José Cruz/ Agência Brasil
Foto: José Cruz/ Agência Brasil  Foto: Dandara Melo Saeb | GOVBA
Foto: Dandara Melo Saeb | GOVBA  Arte ilustrativa / IA
Arte ilustrativa / IA  Foto: Douglas Amaral
Foto: Douglas Amaral  Foto: Amo Animais
Foto: Amo Animais  Imagem gerada por IA
Imagem gerada por IA  Imagem ilustrativa gerada por IA
Imagem ilustrativa gerada por IA  Arte: Tribuna do Recôncavo
Arte: Tribuna do Recôncavo  Image by Michal Jarmoluk from Pixabay
Image by Michal Jarmoluk from Pixabay  Image by Kaufdex from Pixabay
Image by Kaufdex from Pixabay  Imagem de S. Hermann & F. Richter por Pixabay
Imagem de S. Hermann & F. Richter por Pixabay  Foto: Klebe Lobo - Equipe Fred Pontes
Foto: Klebe Lobo - Equipe Fred Pontes  Foto: Marcelo Camargo/ Agência Brasil
Foto: Marcelo Camargo/ Agência Brasil  Foto: Hélio Alves/ Tribuna do Recôncavo
Foto: Hélio Alves/ Tribuna do Recôncavo  Foto: Divulgação
Foto: Divulgação 
 Imagem ilustrativa de Wokandapix por Pixabay
Imagem ilustrativa de Wokandapix por Pixabay  Arte: Divulgação
Arte: Divulgação  Imagem de
Imagem de  Foto: Adelson Menezes
Foto: Adelson Menezes  Foto: Leonardo Rattes/ Saúde GovBA
Foto: Leonardo Rattes/ Saúde GovBA  Arquivo Pessoal
Arquivo Pessoal  Imagem editada de Qui Nguyen Khac por Pixabay
Imagem editada de Qui Nguyen Khac por Pixabay  Foto: Leitora do Tribuna do Recôncavo
Foto: Leitora do Tribuna do Recôncavo  Image by
Image by Imagem ilustrativa | Foto: Hélio Alves/ Tribuna do Recôncavo
Imagem ilustrativa | Foto: Hélio Alves/ Tribuna do Recôncavo  Foto: Luciano Almeida
Foto: Luciano Almeida  Foto: Divulgação
Foto: Divulgação  Foto: Hélio Alves/ Tribuna do Recôncavo
Foto: Hélio Alves/ Tribuna do Recôncavo  Imagem ilustrativa de Couleur por Pixabay
Imagem ilustrativa de Couleur por Pixabay  Fotografia: Telma Galino
Fotografia: Telma Galino  Foto: Reprodução/COPPA
Foto: Reprodução/COPPA  Foto: Fábio Pozzebom/ Agência Brasil
Foto: Fábio Pozzebom/ Agência Brasil  Imagem Ilustrativa | Arquivo: Tribuna do Recôncavo
Imagem Ilustrativa | Arquivo: Tribuna do Recôncavo  Imagem de Cristiano Cardoso por Pixabay
Imagem de Cristiano Cardoso por Pixabay  Imagem ilustrativa | Foto: Hélio Alves/ Tribuna do Recôncavo
Imagem ilustrativa | Foto: Hélio Alves/ Tribuna do Recôncavo  Imagem de adaoaalves por Pixabay
Imagem de adaoaalves por Pixabay  Video
Video 
 PM
PM  Foto: Victor Ferreira/ EC Vitória
Foto: Victor Ferreira/ EC Vitória  Reprodução/ Video
Reprodução/ Video  Imagem de Luk Luk do Pixabay
Imagem de Luk Luk do Pixabay  Foto: Hélio Alves/ Tribuna do Recôncavo
Foto: Hélio Alves/ Tribuna do Recôncavo  Image by 3D Animation Production Company from Pixabay
Image by 3D Animation Production Company from Pixabay  Foto: Itatim News
Foto: Itatim News  Imagem ilustrativa by Free-Photos from Pixabay
Imagem ilustrativa by Free-Photos from Pixabay  Foto: Marcelo Casal Jr./ Agência Brasil
Foto: Marcelo Casal Jr./ Agência Brasil  Foto: Najara Araújo/ Agência Câmara
Foto: Najara Araújo/ Agência Câmara  Divulgação
Divulgação  Foto: Reprodução/ Vídeo/ YouTube/ Mercado Livre Brasil Oficial
Foto: Reprodução/ Vídeo/ YouTube/ Mercado Livre Brasil Oficial  Foto: Luciano Almeido
Foto: Luciano Almeido  Foto: Marcello Casal Jr/ Agência Brasil
Foto: Marcello Casal Jr/ Agência Brasil  Foto: Mateus Pereira/ GOVBA
Foto: Mateus Pereira/ GOVBA  Foto: Eduardo AndradeAscomSDE.jpeg
Foto: Eduardo AndradeAscomSDE.jpeg  Divulgação
Divulgação  Arquivo Pessoal
Arquivo Pessoal  Reprodução: Recôncavo no AR
Reprodução: Recôncavo no AR  FotoS: Reynaldo Felix / AgFpontes / Divulgação.
FotoS: Reynaldo Felix / AgFpontes / Divulgação.  Edição: Tribuna do Recôncavo
Edição: Tribuna do Recôncavo  Imagem de whekevi por Pixabay
Imagem de whekevi por Pixabay  Imagem de MasterTux do Pixabay
Imagem de MasterTux do Pixabay  Arquivo Pessoal
Arquivo Pessoal  Foto: Adam Vidal
Foto: Adam Vidal  Imagem Ilustrativa | Foto: Hélio Alves/ Tribuna do Recôncavo
Imagem Ilustrativa | Foto: Hélio Alves/ Tribuna do Recôncavo  Foto: Marcello Casal Jr/ Agência Brasil
Foto: Marcello Casal Jr/ Agência Brasil  Foto: Divulgação
Foto: Divulgação 
 Divulgação
Divulgação  Foto: Divulgação
Foto: Divulgação  Foto: Jamile Amine / Saúde GovBA
Foto: Jamile Amine / Saúde GovBA  Imagem ilustrativa by Esi Grünhagen from Pixabay
Imagem ilustrativa by Esi Grünhagen from Pixabay